
October 30, 2020
Weizmann Institute of Science researchers have found that gut microbes and host cells jointly contribute to the progression of acute liver failure – a mostly incurable disease – yet also gave clues to treatment.
Acute liver failure is a devastating, rapidly progressing disease that results in death in 80% of cases, unless an emergency liver transplant is performed. In the developed world, its leading cause is a substantial overdose of acetaminophen, also known as paracetamol.
In a study published in Nature Medicine, Weizmann researchers from the labs of Professors Eran Elinav and Ido Amit in the Immunology Department have, by using mouse models of acute liver failure, discovered three new subsets of liver cells that orchestrate the development of this condition.
The scientists also uncovered signals – from the gut microbiome as well as the diseased liver – that jointly activate these cells, and showed that selectively blocking these signals and depleting the microbiome led to marked improvement in liver function and prolonged survival in mice.
An analysis of liver tissue from human patients with acute liver failure revealed a molecular pattern strikingly similar to the one identified in mice in the study, raising hopes that the findings in mice may in the future be translated into a treatment for humans.
Dr Aleksandra Kolodziejczyk, a postdoctoral fellow in Elinav’s lab, led this project in collaboration with other scientists at the Weizmann Institute of Science and Dr Amir Shlomai of the Liver Institute, Rabin Medical Center.
Kolodziejczyk and her colleagues began their exploration by creating gene expression profiles of 45,000 individual mouse liver cells, ultimately generating a comprehensive liver cell atlas in conditions of health and acute liver failure. The scientists identified 49 cell subsets, of which three new subsets – among stellate, endothelial and Kupffer cells – became abnormally activated as the acute liver failure progressed in the mice.
These previously undescribed cell subsets secreted a large variety of substances that attracted immune cells from outside the liver, which then contributed to its damage. All three new cell subtypes shared a characteristic expression pattern of 77 genes – a pattern controlled by the same regulatory protein, the transcription factor MYC – which suggested that these cells may be activated through a common program.
The researchers suspected the newly uncovered activation pathway could be regulated by signals from the gut microbiome. This makes anatomical sense, as the gastrointestinal tract drains into the liver though a large network of veins, directly exposing the liver to substances produced in the gut and by its microbes. When the scientists depleted the microbiome of the mice by administering wide-spectrum antibiotics, symptoms of liver failure were alleviated. Moreover, when they induced acute liver failure in germ-free mice, which lack a microbiome, the condition was much less severe than in regular mice. Further studies of mice with and without a gut microbiome revealed that during acute liver failure, distinct molecules generated by the microbiome accumulate in the liver, where they activate the MYC protein in the three liver cell subtypes that contribute to liver damage. In the absence of a microbiome, MYC activation was attenuated, leading to reduced liver damage.
Kolodziejczyk then worked out the molecular details of MYC activation. She found that the molecules coming from the microbiome activate the MYC program through surface receptors on the three cell subtypes that she had identified earlier as aggravating the liver failure. She also found that the MYC program was activated in the same manner – that is, through the same receptors on the three cell subtypes – by signals coming from liver cells damaged by paracetamol.
When the mice were genetically depleted of functioning receptors, given drugs that blocked MYC or otherwise had the signals between these receptors and MYC interrupted, they no longer developed acute liver failure and their survival was extended. Gene expression analysis of individual cells showed that in the treated mice, the three newly identified cell subtypes were no longer abnormally activated, and this reduced both the immune cell infiltration and the resultant liver damage.
Finally, the researchers teamed up with Dr Shlomai to analyze liver samples from patients with acute liver failure and to compare them with samples from healthy liver donors. Those from the patients – but not from healthy donors – were characterised by robust MYC activation that was similar to the one observed in mice.
These results raise the possibility that blocking the MYC program by drugs, coupled with microbiome modulation, may prove to be a potential treatment for acute liver failure.
“Our findings provide a first step towards achieving a comprehensive understanding of how the microbiome interacts with the host in contributing to acute liver failure,” Elinav said.
“Such knowledge could lead to a new treatment option for this cureless and devastating disorder.”
Also taking part in the study were Dr Sara Federici, Dr Niv Zmora, Dr Gayatree Mohapatra, Dr Mally Dori-Bachash, Shanni Hornstein, Dr Avner Leshem and Dr Hagit Shapiro of the Elinav lab; Dr Tomer Meir Salame of Weizmann’s Life Sciences Core Facilities; Professor Alon Harmelin of Weizmann’s Veterinary Resources Department; Dr Debby Reuveni and Dr Ehud Zigmond of the Tel Aviv Sourasky Medical Center; and Dr Ana Tobar of the Rabin Medical Center.
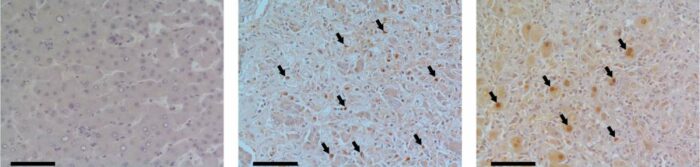
Human liver tissue under a microscope. Compared to healthy donors (left), tissue samples from people with acute liver failure (center, right) reveal a significant increase in levels of the MYC protein (brown) in the cell nuclei (some of which are indicated by arrows)
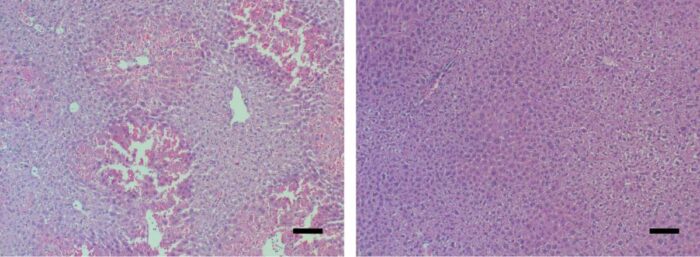
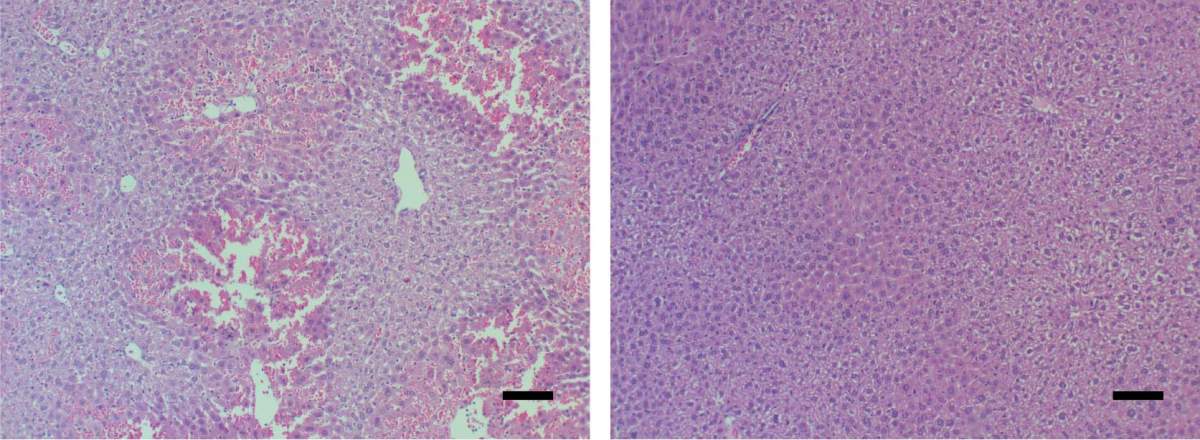
Mouse liver tissue under a microscope. Signs of acute liver failure (left) disappeared (right) after the mice received a MYC-blocking drug





